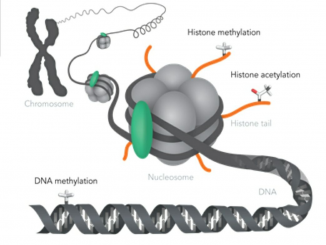
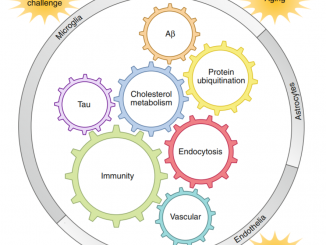
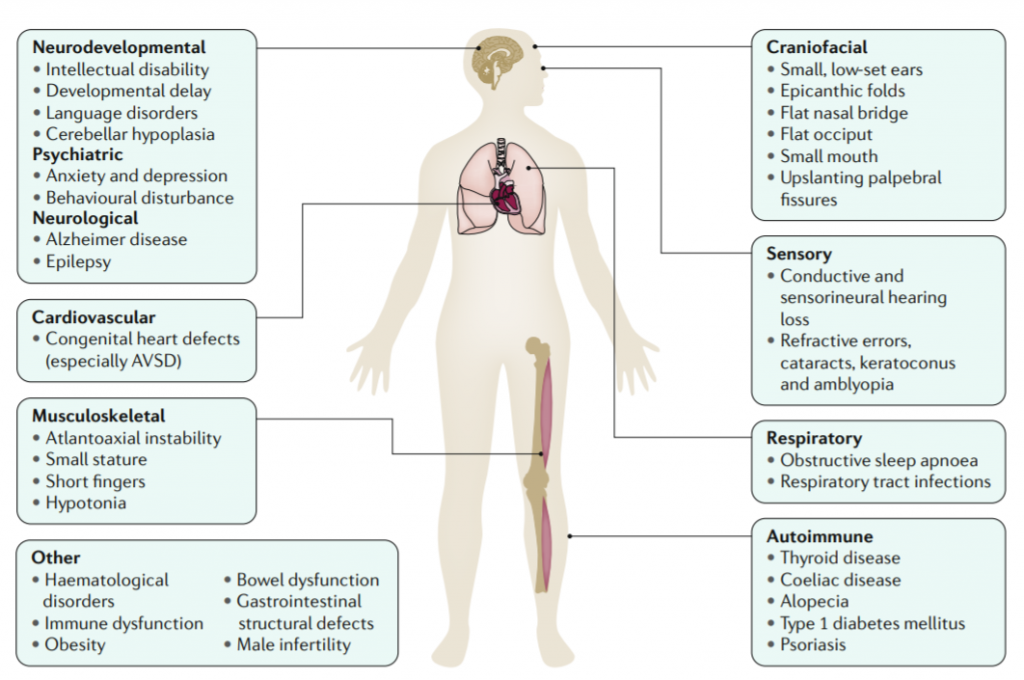
خلاصه مقاله مروری نیچر در مورد سندرم داون و چشم اندازهای درمان در سندرم داون
وجود یک کروموزوم اضافی ۲۱ در انسان منجر به مجموعهای از ویژگیهای بالینی میشود که به نام سندروم داون شناخته شده است .سندروم داون در زمره پیچیده ترین حالات ژنتیکی است که با حیات بعد از تولد سازگار است این بیماری همچنین شایع ترین آنوپلوییدی اتوزومی سازگار با حیات در انسان است. بیشتر مدلهای مدل موشی سندرم داون که شامل همه یا قسمتی از کروم ۲۱ و یا مناطق مشابه کروموزوم ۲۱ در انسان است اطلاعات با ارزشی در مورد سه تایی شدن ژن ها و یا گروهی از ژن ها و در نتیجه بسیاری از علائم بالینی آنها در سندرم داون ارائه می کند.

در مجموع تحلیل این یافته ها مشکل است زیرا که بیشتر از ۲۰۰ ژن کد کننده پروتئین بر روی کروموزوم ۲۱ وجود دارد و آنها می توانند اثرات مستقیم و یا غیر مستقیم در هموستاز سلول ها و بافت ها ارگان ها و سیستم ها داشته باشند.
هرچند این پیچیدگی چلشهای مهمی را در فهم زیربنای مولکولی هر کدام از این ویژگی های بالینی ایجاد می کند با این وجود فرصتی برای بهبود فهم ما از مکانیزم های ژنتیکی در زمینه تکامل و عملکرد بسیاری از انواع سلول ها و بافت ها ارگان ها و سیستم ها هم فراهم می کنند .
از زمان توصیف نخستین بار تریزومی ۲۱ تاکنون ما دانش ما در مورد ناتوانی ذهنی و ریسک فاکتورهای بیماریهای قلبی مادرزادی بسیار افزایش یافته است.
وقوع کمتر تومورهای بافت های سخت در افراد مبتلا به سندروم دان به شناسایی ژن هایی بر روی کروموزوم ۲۱ که زمانی که بیان بیشتری شوند در برابر سرطان حفاظت کننده هستند کمک می کند. از طرفی وقوع فراگیر هیستوپاتولوژی آلزایمر و شیوع بالا دمانس در سندروم دان به ما به فهم بیشتر و درمان آلزایمر کمک می کند. برای آزمون های بالینی در زمینه ناتوانی های ذهنی در سندروم دان رویکرد های جدیدی از درمانهای مداخلهای بر مبنای شناخت ما از پاتوفیزیولوژی مولکولی سندروم دان اکنون فراهم شده است و برای آینده امید هایی وجود دارد.